The first crystal structure of a G-protein-coupled receptor in complex with an arrestin protein provides insight into how the signalling pathways activated by these receptors are switched off through desensitization.
G-protein-coupled receptors (GPCRs) play an essential part in mediating signalling in the cells of many organisms. This process is primarily controlled by activation-dependent interactions of GPCRs with three protein families: heterotrimeric guanine-nucleotide-binding proteins (G proteins), GPCR kinases (GRKs) and arrestins. Until now, the only complete structure1 of a GPCR complex was that of the β2-adrenergic receptor bound to the G protein Gs, solved in 2011. In a paper published on Nature's website today, Kang et al.2 present the second structure of a GPCR complex, in this case the GPCR rhodopsin bound to arrestin-1.
The binding of a GPCR to a G protein results in the activation and subsequent regulation of downstream effector enzymes that modulate levels of 'second messenger' molecules such as cyclic AMP and calcium. By contrast, the interaction of a GPCR with a GRK promotes phosphorylation of the receptor, which in turn facilitates arrestin binding. This turns off G-protein signalling, a process called desensitization, and promotes cellular internalization of the receptors and arrestin-mediated signalling. The dynamics of these protein–protein interactions are complex and incompletely understood.
The best-characterized GPCR signalling pathway regulates the process of phototransduction in rod cells in the retina of the eye. This pathway involves rhodopsin, the G protein transducin and the effector enzyme cGMP phosphodiesterase (Fig. 1a). Phototransduction is highly regulated by a GRK (GRK1) and an arrestin (arrestin-1); mutations in either of these proteins can lead to a visual defect called Oguchi disease. Rhodopsin was the first GPCR to be crystallized in its basal state3 as well as in various activated conformations4, 5, 6. Although some structural insight into the binding of G proteins5, 6 and arrestins7 to rhodopsin has been gained by using peptides co-crystallized with the receptor, Kang and colleagues' structure of the full protein complex is a significant advance.
Figure 1: The phototransduction pathway.
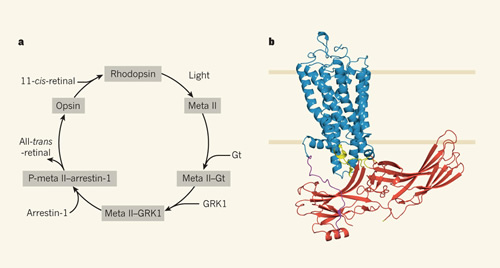
a, The signalling pathway involving the G-protein-coupled receptor rhodopsin is initiated when light activation induces rhodopsin to form meta II rhodopsin, which interacts with the G protein transducin (Gt) to activate the effector enzyme cGMP phosphodiesterase. This pathway ultimately leads to a rapid visual response in rod cells. Meta II is then phosphorylated by the protein kinase GRK1 to yield P-meta II, which promotes interaction with the protein arrestin-1 to preclude further binding of Gt. P-meta II recycles by losing its bound chromophore (all-trans-retinal) to become P-opsin (not shown), which promotes arrestin-1 dissociation and dephosphorylation of the receptor. Opsin then binds the chromophore 11-cis-retinal to yield rhodopsin. Each of the individual proteins in this pathway (rhodopsin, meta II, opsin, Gt, GRK1 and arrestin-1) have been crystallized. b, Kang et al.2 provide the first crystal structure of a protein complex in this pathway, that of activated rhodopsin (blue) bound to arrestin-1 (red). Sections of arrestin-1 involved in the interface with rhodopsin are shown in yellow. The carboxy terminus of rhodopsin, which is phosphorylated by GRK1, is shown in purple. Rhodopsin is depicted in a phospholipid bilayer.
Solving the X-ray structure of a rhodopsin–arrestin complex proved challenging and required a team of 72 investigators across 25 institutions, who used various tricks to obtain diffractable crystals. First, the two proteins were mutated to aid the formation of active conformations. For rhodopsin, these mutations (E113Q and M257Y) yielded a conformation that was constitutively active even in the absence of a bound chromophore (a colour-determining chemical group, such as all-trans-retinal, which typically activates rhodopsin). For arrestin, the researchers mutated three adjacent amino-acid residues in a region that stabilizes the basal conformation and largely overcomes the need for the receptor to be phosphorylated to bind arrestin8.
The authors were unable to generate a stable rhodopsin–arrestin binary complex (one formed through noncovalent binding). Instead, they purified and crystallized a fusion protein in which arrestin was fused to the carboxy terminus of rhodopsin through a 15-residue linker. This fusion protein also included the enzyme T4 lysozyme at the amino terminus of rhodopsin — this facilitates crystallization without altering the structure of the complex. Finally, because the crystals were small and diffracted to 6–8 ångström in synchrotron experiments, the authors used serial femtosecond X-ray laser crystallography, performed at the SLAC National Accelerator Laboratory in Menlo Park, California, to capture diffraction data for the crystals. This provided enough data to solve a structure of the complex with resolution limits of 3.3–3.8 Å.
The structure reveals multiple points of contact between rhodopsin and arrestin, as well as structural changes in both proteins (Fig. 1b). The primary interface between the proteins involves the finger loop of arrestin (which connects β-strands V and VI in arrestin and adopts an α-helical conformation when bound), which interacts with three regions of rhodopsin: intracellular loop (ICL) 1, the N-terminal region of helix 8, and the C-terminal region of transmembrane (TM) 7. Additional interactions include several arrestin loops: the middle and lariat loops bind to ICL2 on rhodopsin, the back loop binds to TM5, and β-strand VI binds to TM5, TM6 and ICL3.
The authors' basic model proposes that rhodopsin uses multiple structural elements including TM7 and helix 8 to initially recruit arrestin, resulting in a rotation of approximately 20° between the N and C domains of arrestin; this opens a cleft to accommodate rhodopsin's ICL2. Indeed, a similar rotation between the N and C domains has been observed in a preactivated truncated form of arrestin-1 and in β-arrestin-1 (also known as arrestin-2) when bound to a phosphorylated receptor peptide. Kang et al. extensively validated their rhodopsin–arrestin structural model by using double electron–electron resonance, hydrogen–deuterium exchange mass spectrometry, cell-based rhodopsin–arrestin interaction assays, and site-specific disulfide cross-linking experiments.
Kang and colleagues' study provides insight into the interactions between GPCRs and arrestins, but much remains to be learned. We clearly need the structures of more GPCR complexes with G proteins, arrestins and GRKs, including complexes of such proteins with the same GPCR — for example, the β2-adrenergic receptor in complex, separately, with Gs, GRK2 and β-arrestin-1, or rhodopsin in complex with transducin and GRK1 to complement the arrestin-1 structure. Such studies will reveal whether these three classes of protein have specific preferences for particular receptor conformations, and should facilitate the development of compounds that might serve as selective modulators of specific GPCR signalling pathways. This would be an important step in helping to treat the many diseases that are mediated by GPCR signalling pathways, which are currently the target for approximately 40% of the pharmaceuticals on the market.