Dopamine (DA) is one of the major neurotransmitters in the human central and peripheral nervous systems, participating in numerous important physiological functions through dopamine receptors, which belong to the seven transmembrane G protein-coupled receptor (GPCR). Imbalances and alterations in dopamine signaling are relevant to many brain disorders, including Parkinson's disease, schizophrenia, and Huntington's disease. As one important subtype of the dopamine receptors, D3R has become a promising target of drug research for Parkinson's disease, drug addiction, and schizophrenia. The development of selective drugs for dopamine receptor subtypes is essential for better efficacy and few side effects. However, the molecular details that how D3R/D2R ligands, including PD128907, pramipexole, and rotigotine, exhibit higher preference for D3R are still unclear, thus limiting the understanding of mechanisms and ability to design better drugs targeting dopamine receptors.
In a study published in Molecular Cell on Feb. 5, scientists from the Shanghai Institute of Materia Medica (SIMM) of the Chinese Academy of Sciences, Zhejiang University, and University of North Carolina Chapel Hill, determined two cryo-electron microscopy (cryo-EM) structures of D3R-Gi in complexes with selective ligands PD-128907 and pramipexole, the latter is an approved drug for treatment on Parkinson's disease and Willis-Ekbom disease.
These structures revealed, for the first time, the molecular secrets of D3R binding to its agonists at atomic level, and offer opportunities for drug optimization and discovery targeting this physiologically important receptor.
Both pramipexole and the small molecule agonist PD-128907 activate D2R and D3R, and show superior selectivity for the D3R subtype over D2R and D4R. Comparing the structural details of the D3R receptor binding to two agonists, pramipexole and PD-128907, respectively, the research team found that the two small molecule agonists have distinct characteristics in their binding to the receptor; comparative analysis with the activation structures of D2R and D4R revealed that the spatial position of the histidine at position 6.55 on TM6 in the binding pocket is the key to determine the ligand's selectivity for subtype of dopamine receptors D2R, D3R and D4R.
Meanwhile, the study also explored the mechanism of dopamine receptor subtypes coupling downstream Gs and Gi selectivity and found that three position-specific differential residues on the receptor TM6 (positions 6.31, 6.36, and 6.38) were the determinants of selective coupling of different dopamine receptor subtypes to Gs or Gi. The study further found that the mode of D3R coupled to Gi is similar to that of other Gi-coupled GPCRs through highly electrostatic interaction types. Furthermore, the structures of D3R-Gi also revealed the molecular basis for the preference of D3R couples to Go. Structural observations of D2R and D3R revealed that the TM6 of D3R is more rigid than D2R, resulting in a smaller intracellular pocket, whereas Go proteins have relatively smaller amino acid side chains than Gi, and thus D3R exhibits more preferential binding to Go protein.
In summary, these findings elucidate in detail the molecular mechanism of selective recognition of ligand by D3R receptor, activation of D3R receptor by selective recognition, and coupling to G protein at near-atomic resolution and also provide important structural models for the development of selective drugs targeting dopamine receptors.
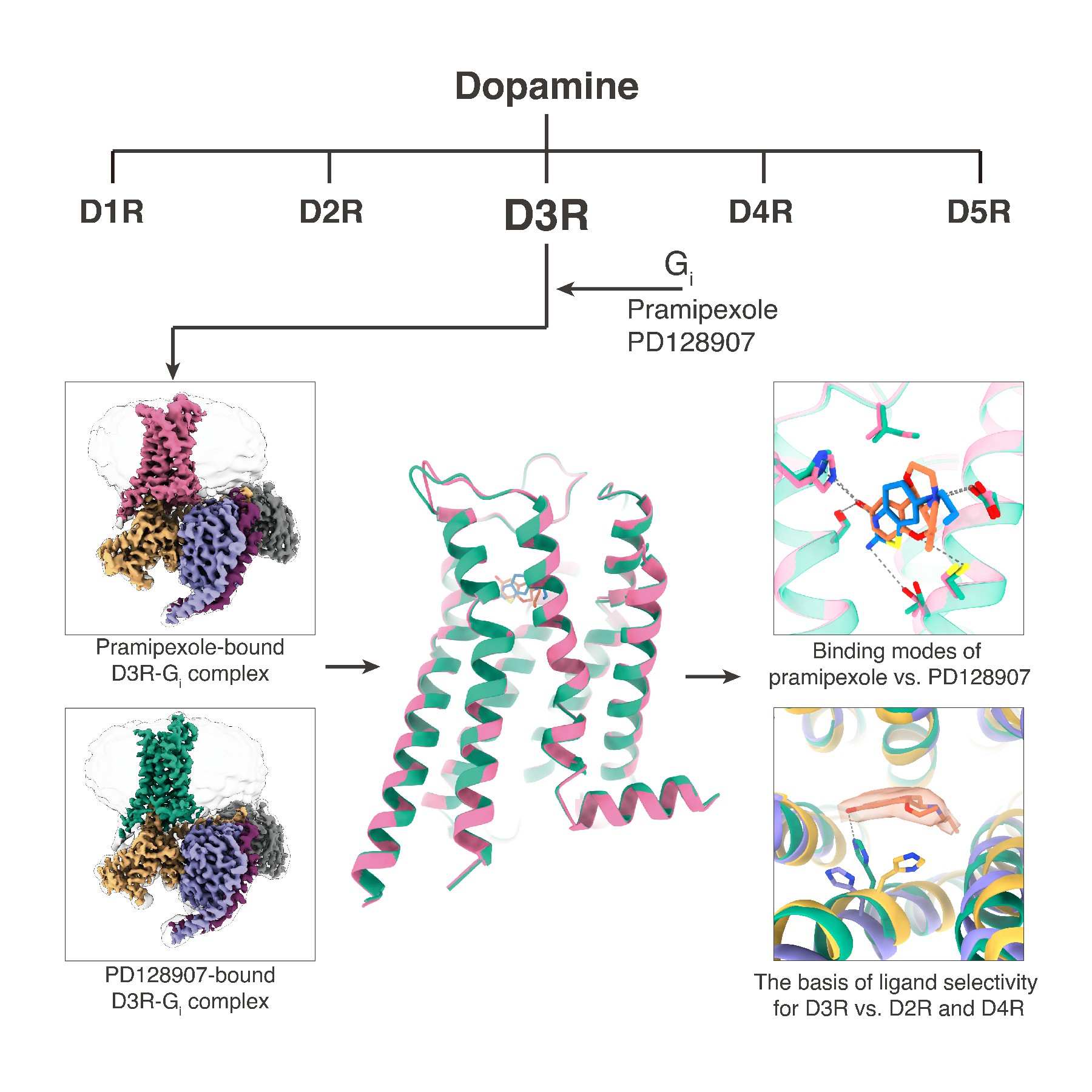
Illustrated representation of the dopamine receptor D3R-Gi complexes and the binding specificity of agonists. (Image by Peiyu Xu)
Link to article: https://www.cell.com/molecular-cell/fulltext/S1097-2765(21)00003-4
Contact: H. Eric Xu, eric.xu@simm.ac.cn