


Efficient Prediction of Relative Ligand Binding Affinity in Drug Discovery
Update time:Author:
Lead optimization in drug discovery is a challenging process that heavily relies on hypotheses and the experience of medicinal chemists. This often leads to uncertain outcomes and inefficiency. Furthermore, the process is time-consuming and requires significant resources. Therefore, the introduction of artificial intelligence (AI) predictive tools to accelerate this process would be highly valuable in the field of drug discovery. In silico methods, such as free energy perturbation (FEP) and molecular mechanics generalized born surface area (MM-GB/SA), have already proven useful in lead optimization by calculating binding free energy. However, their complex preparation process, limited molecule throughput, and restricted allowance for changes between molecules have hindered their routine usage. Recently, there have been proposals for fast and user-friendly deep learning models for binding affinity prediction, but their accuracy is still not satisfactory. In conclusion, there is an urgent need to develop an efficient and accurate in silico predictive tool to guide lead optimization.
In a study published in Nature Computational Science on October 19, a team of researchers led by Prof. ZHENG Mingyue from the Shanghai Institute of Materia Medica (SIMM) of the Chinese Academy of Sciences developed a pairwise binding comparison network (PBCNet). This network predicts the relative binding affinity among congeneric ligands by using a physics-informed graph attention mechanism with a pair of protein pocket-ligand complexes as input. PBCNet demonstrates practical value in guiding structure-based drug lead optimization with its speed, precision, and ease-of-use.
To validate the performance of PBCNet in terms of ranking ability and accuracy, ZHENG's group used two held-out sets provided by Schrodinger, Inc. and Merck KGaA. These sets comprised over 460 ligands and 16 targets. Transfer learning was applied in their work, involving pretraining models on large-scale datasets and fine-tuning them for tasks with limited data. This approach specifically improved the performance of the models on the tasks. Benchmarking results obtained from the test data showed that pretrained PBCNet outperformed Schrodinger's Glide, MM-GB/SA, and four recently reported deep learning models (DeltaDelta, Default2018, Dense, and PIGNet). Furthermore, with a small amount of fine-tuning data (2-10 ligands with known binding activity), PBCNet achieved performance comparable to that of Schrodinger's FEP+, which is considered the standard computational lead optimization method in the pharmaceutical industry. The researchers also tested whether PBCNet could efficiently identify key high-activity compounds in a real-world lead optimization scenario. They used a benchmark consisting of nine recently published chemical series and compared the order of model selection to the experimental order of synthesis. The evaluation demonstrated that, after using PBCNet, the tested lead optimization projects were accelerated by approximately 473% while resource investment was reduced by an average of 30%.
Based on these results, PBCNet has immediate practical value in guiding lead optimization projects. Additionally, there is a free academic web service available at https://pbcnet.alphama.com.cn/index that utilizes PBCNet to predict ligand binding affinity. AI has become increasingly important in solving scientific problems by incorporating domain-specific knowledge into its models. PBCNet exemplifies this approach by integrating physical and a priori knowledge into its modeling process.
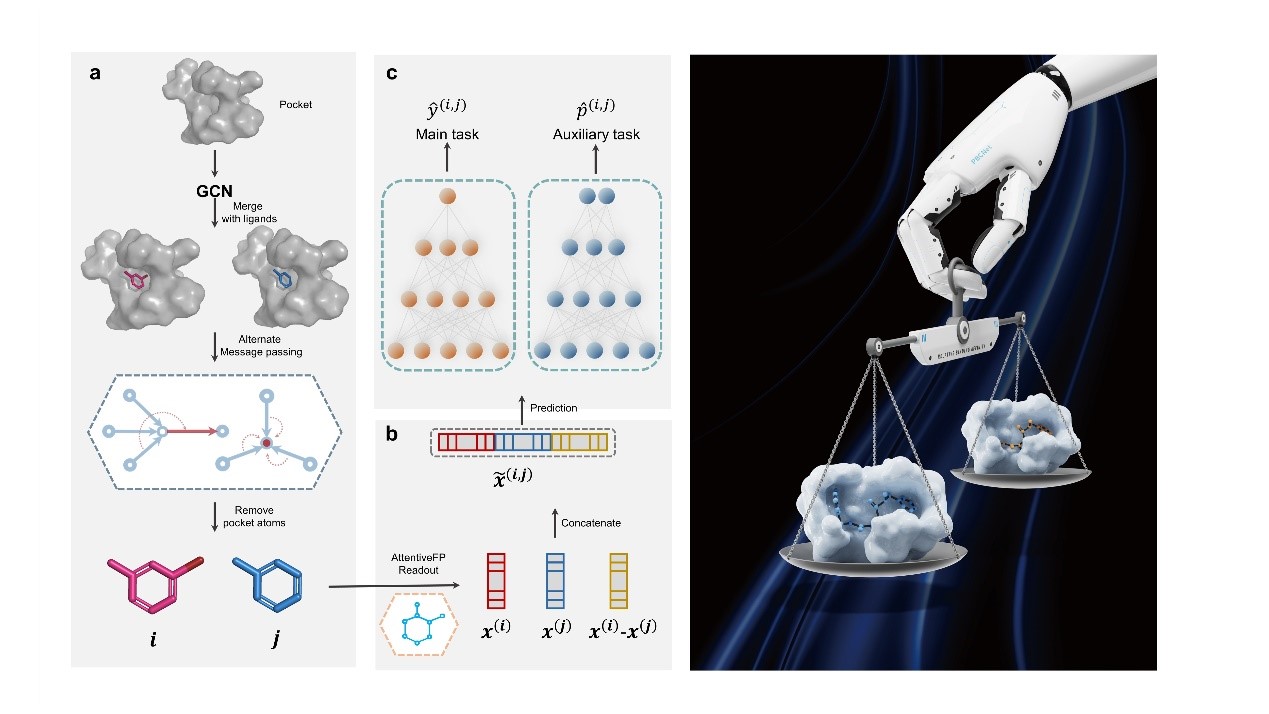
The PBCNet framework (left) The Schematic illustration of PBCNet’s working principle (right)
(image by ZHENG Mingyue's laboratory at SIMM)
DOI number: https://doi.org/10.1038/s43588-023-00529-9
URL: https://www.nature.com/articles/s43588-023-00529-9
Key words: Lead optimization, Structure‐based drug design, Deep learning
Contact:
DIAO Wentong
Shanghai Institute of Materia Medica, Chinese Academy of Sciences
E-mail: diaowentong@simm.ac.cn