


Researchers Develop Annotation Approach for Endogenous Retroviruses and Reveal Their Divergent Evolution Across Species
In a study published in Science Advances, a team of researchers led by CHEN Xun from the Institute, Guillaume Bourque and Fumitaka Inoue from Kyoto University, Japan developed a novel phylogeny-based approach for the classification and annotation of endogenous retroviruses (ERVs). By integrating phylogenetics, massively parallel reporter assays (MPRAs) and multi-omics techniques, they revealed differences in endogenous retrovirus sequences and their regulatory functions at the single-base resolution across various species during evolution.
The human virome encompasses infectious viruses, integrated exogenous viruses (like Hepatitis B virus), and endogenous retroviruses (ERVs). Endogenous retroviruses are a major type of transposable elements, constituting approximately 8% of the human genome. They originate from ancient retroviral infections and were endogenized in the genome millions of years ago. There are many types of endogenous retroviruses, such as HERV-E, HERV-K, and HERV-H. Based on sequence variations, they can be classified into over 500 subfamilies.
A full-length endogenous retrovirus typically contains three core gene domains – gag, pol, and env - flanked by long terminal repeats (LTRs) crucial for regulation. Most ERVs lost their function due to the accumulated mutations or rearrangements during evolution, leading to their historical designation as genomic "junk DNA." However, recent research has shown that endogenous retroviruses, particularly their LTR sequences, harbor numerous transcription factor binding sites. They can act as cis-regulatory elements, modulating the expression of nearby genes and involving in regulatory networks including innate immunity. Thus, endogenous retroviruses play critical roles in various human diseases including cancers, developmental disorders, and infectious diseases.
Accurate annotation of endogenous retrovirus sequences is fundamental to understanding their function and evolution. However, current methods relying primarily on sequence alignment were limited, resulting in numerous errors in human genome ERV annotation. To address this challenge, the researchers first developed a novel strategy for transposable element annotation. This phylogeny-guided approach integrates and re-annotates sequences that are evolutionarily close but may be misclassified into different subfamilies. Applying this approach to 76 young endogenous retrovirus subfamilies in human, they successfully corrected approximately one-third of the sequence annotations in 26 subfamilies.
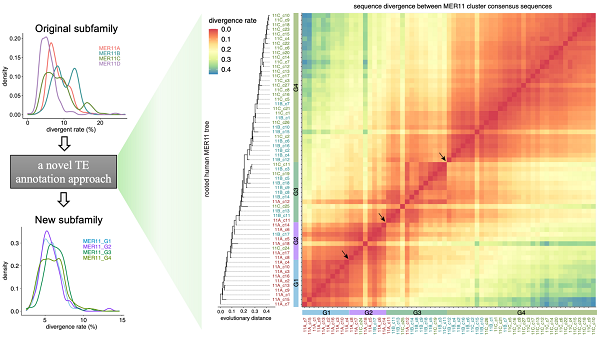
Figure 1. A phylogeny-based transposable element annotation approach. (Image by CHEN Xun)
Focusing on the MER11 family as an example, the team combined phylogenetic analysis with epigenomics data to uncover extensive annotation errors within the known MER11A/B/C subfamilies. By applying the approach, they newly annotated these MER11 sequences into four new subfamilies: MER11_G1, G2, G3, and G4, which could well re-arrange the epigenetic states across the MER11 sequences and trace their evolutionary history.
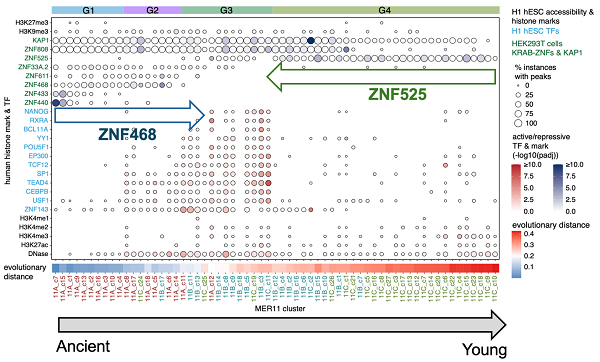
Figure 2. The evolution of epigenetic profiles revealed by the new MER11 subfamilies. (Image by CHEN Xun)
The team then used MPRAs technology to experimentally validate the transcriptional regulatory activity (promoter/enhancer function) of over 7,000 MER11 sequences from human, chimpanzee, and macaque genomes. This analysis revealed key transcription factor binding motifs like SOXs and their evolutionary dynamics at single-base resolution. Notably, they identified a SOX-related motif specific to humans and chimpanzees in evolutionarily young MER11 sequences. This functional motif arose during primate evolution through a single base-pair deletion event that significantly enhanced its regulatory activity.
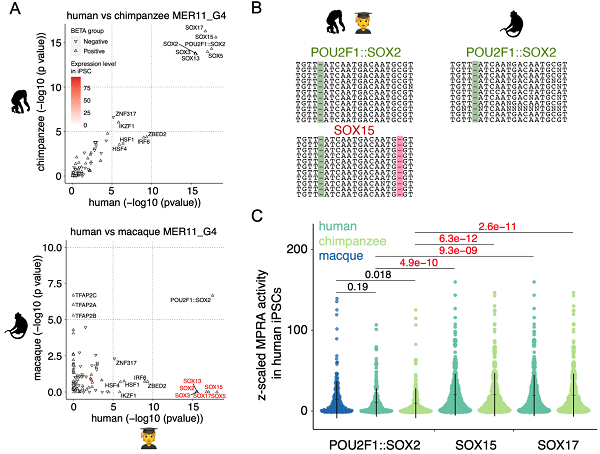
Figure 3. The gain of functional motifs due to nucleotide changes during separate expansions of endogenous retroviruses in primate lineages. (Image by CHEN Xun)
In summary, this study establishes a comprehensive methodology for investigating the classification, annotation, evolutionary history, and biological functions of transposable elements like endogenous retroviruses. Using this approach, researchers could trace the co-evolutionary trajectory of specific endogenous retrovirus sequences and their functional impacts. Furthermore, the precise annotation will significantly enhance the downstream sequence and functional analyses. This provides a powerful tool for systematically investigating the biological roles and evolution of endogenous retroviruses in tumorigenesis, developmental regulation, immune-related diseases, and beyond.
Looking ahead, the research team aims to further integrate phylogenetics, multi-omics technologies, and artificial intelligence (AI) approaches to dissect endogenous retroviruses' complex biological functions, to explore hidden targets and their critical roles in human immune system.
Link: https://doi.org/10.1126/sciadv.ads9164
Contact:
DIAO Wentong
Shanghai Institute of Materia Medica
E-mail: diaowentong@simm.ac.cn