


Scientists Unveil Mechanisms under Ligand Selectivity and G Protein Coupling Selectivity via Dopamine Receptors D1R and D2R Signaling Complexes Structures
Dopamine, a catecholamine neurotransmitter with important functions for both the central (CNS) and peripheral (PNS) nervous systems, participates in advanced brain functions like rewarding and pleasure feeling. Dopaminergic functions are mainly mediated by a family of five G-protein-coupled receptors, which can be divided into two groups: the D1-like and the D2-like receptors. The D1-like group, including D1R and D5R, primarily couple to the stimulatory G protein Gs, while the D2-like group, including D2R, D3R and D4R, primarily couple to the inhibitory G protein Gi/o. Among the five dopamine receptors (DRs), D1R and D2R are the most abundant receptors in CNS. Aberrant D1R and D2R signaling has been associated with many neuropsychiatric diseases such as Parkinson’s Disease (PD), schizophrenia and drug abuse and autism. Insight into their signaling is essential to understand both therapeutic and side effects of dopaminergic drugs. However, lack of D1R structure and detail comparison of ligand binding between D1R and D2R largely impede the selective drug discovery targeting D1R and D2R.
To resolve these scientific questions, scientists from the Shanghai Institute of Materia Medica (SIMM) of the Chinese Academy of Sciences, together with multiple groups, determined four near-atomic structures of D1R-Gs and D2R-Gi signaling complexes activated by divergent agonists using cryo-electron microscopy (cryo-EM) method. These ligands mainly include D1R/D5R selective agonists SKF81297, SKF83959 and DRs non-selective agonists apomorphine and bromocriptine. The structural data combined with functional studies reveals both the ligand selectivity and G protein coupling selectivity of D1R and D2R and also the structure determinants of biased agonism of SKF83959 to D1R. The research has been currently published online in Cell.
In this study, Prof. H. Eric Xu, cooperating with several other teams, provides the first insights into the D1R structure and its ligand binding pocket topology and the G protein coupling details. Structure comparison shows that the D1R structure displays high similarity with β2AR in active state even in the conserved PIF motif and DRY motif. Further mutagenesis studies reveal that non-conserved residue V317 plays an important role in catecholamine neurotransmitter selectivity between D1R and β2AR. SKF83959 is a G protein biased agonist of D1R, the study also reveals that residues F288, F289 and V317 in D1R TM region and the additional methyl group of SKF83959 compared to SKF81297 play vital role in G protein biased activity of SKF83959 toward D1R.
Moreover, researchers found, through multiple comparison, that the ECL2 region is a significant regulator of ligand selectivity of D1R and D2R. In addition, the differences in whole topology of ligand binding pockets of D1R and D2R, especially the different conformation of TM6 and the non-conserved residue K81 in D1R, contribute to the ligand selectivity of the two receptors.
Finally, scientists further suggests the differences in D1R and D2R, the outward movement of TM6 and the extension of TM5 in the cytoplasmic side of D1R relative to D2R, serve as a basis for G protein-coupling specificity. These structural differences allow D1R to primarily couple with Gs but not Gi, and for D2R to couple Gi but not Gs.
“We present the first view of human D1R, an important receptor in dopaminergic system, and this study provides unprecedented structural insights into the pharmacology and signaling of D1R and D2R, together with D3R structures published last week in Molecule Cell, these structures inspire a brand new access to discovering safer and more efficient drugs treating CNS diseases, like PD and schizophrenia.” Prof. Xu said.
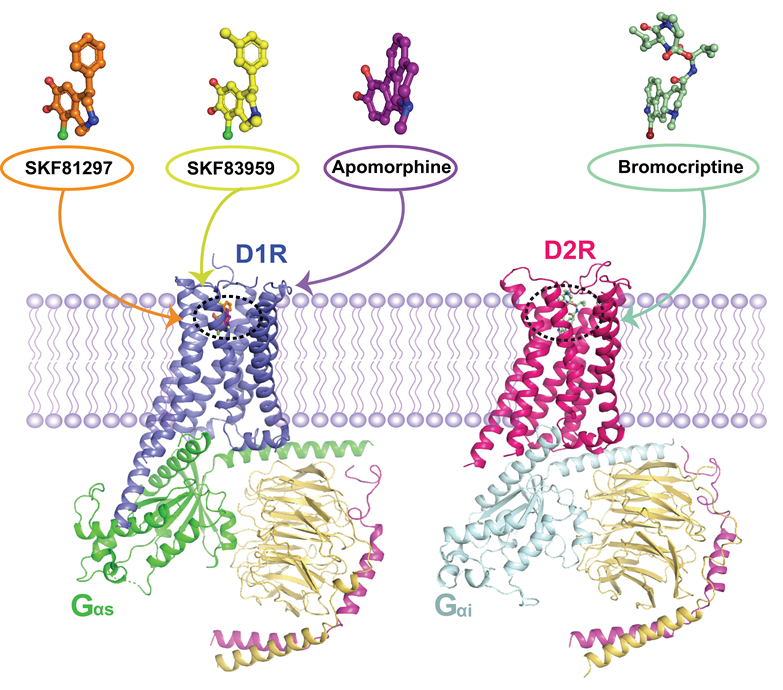
Structures of D1R-Gs and D2R-Gi complexes. (Image provided by H. Eric Xu’s group)
Link to article:
Contact:
Youwen Zhuang, stevenzh_xulab2014@simm.ac.cn
H. Eric Xu, eric.xu@simm.ac.cn