


New AI tool efficiently deconvolutes compound-protein interactions
Compounds and proteins are the two most fundamental entities in drug discovery. Modeling their interactions is crucial for drug discovery and represents a shared goal among researchers. Although no universal computational method currently exists to predict and explain all compound-protein interactions (CPIs), researchers can contribute to building a comprehensive CPI map by leveraging various biological data from different perspectives.
Recent advances in high-throughput transcriptomic screening have opened new avenues for drug discovery. Perturbation transcriptomics, which reflects cellular transcriptomic responses to perturbations, links key entities (compounds) with omics data. This approach provides intuitive results of how compounds affect subjects (single cells, cell lines, patients), offering a fresh perspective for decoupling CPIs.
In a study published in Cell Genomics, a research team led by ZHENG Mingyue, ZHANG Sulin, and LI Xutong from the Shanghai Institute of Materia Medica (SIMM) of the Chinese Academy of Sciences, developed an AI tool called PertKGE. This tool deconvolutes CPI from perturbation transcriptomics using knowledge graph embedding.
PertKGE built upon existing biomedical knowledge graphs but employed a novel strategy. The key innovation lay in constructing a biologically meaningful knowledge graph that broke down genes into DNAs, messenger RNAs (mRNAs), long non-coding RNAs (lncRNAs), microRNAs (miRNAs), transcription factors (TFs), RNA-binding proteins (RBPs), and other protein-coding genes. This approach allowed PertKGE to capture various fine-grained interactions between genes, simulating post-transcriptional and post-translational regulatory events in biological systems. PertKGE then used the knowledge graph embedding method DistMult to project all entities into a semantically rich hidden space, enabling deconvolution of CPIs from perturbation transcriptomics.
Researchers extensively benchmarked existing approaches under two critical "cold-start" settings: inferring binding targets for new compounds and conducting virtual ligand screening for new targets. PertKGE outperformed all traditional methods and deep learning approaches. They further demonstrated the pivotal role of incorporating multi-level regulatory events in mitigating representational biases.
Notably, combining PertKGE with phenotype-based and target-based drug discovery led to two significant findings. The first was the identification of ectonucleotide pyrophosphatase/phosphodiesterase-1 (ENPP1) as target responsible for the unique anti-tumor immunotherapy effect of tankyrase inhibitor K-756. The second was the discovery of five novel hits targeting the emerging cancer therapeutic target, aldehyde dehydrogenase 1B1.
These results strongly suggested that PertKGE can help pharmacologists accelerate drug discovery. Looking ahead, PertKGE is expected to integrate more regulatory events, further enhancing its predictive performance and expanding its application to the analysis of other perturbation omics data.
DOI: doi.org/10.1016/j.xgen.2024.100655
Link: https://www.cell.com/cell-genomics/fulltext/S2666-979X(24)00266-0
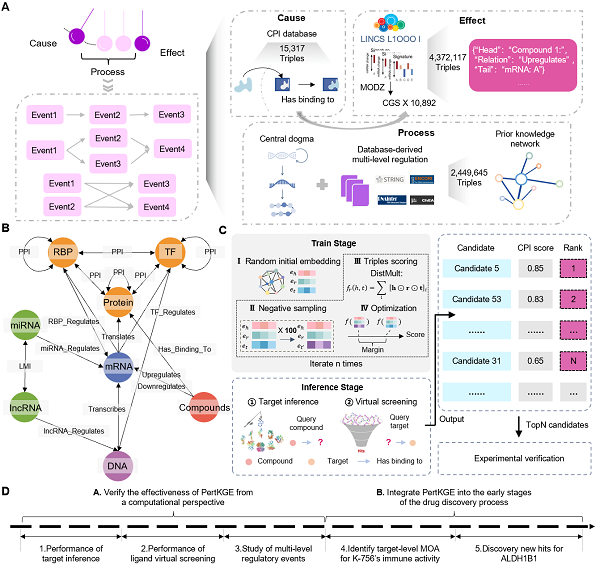
Overview of PertKGE workflow. (Image by ZHENG’s lab)
Contact:
JIANG Qingling
Shanghai Institute of Materia Medica, Chinese Academy of Sciences
E-mail: qljiang@stimes.cn