


New AI Model for Reliable and Accurate Protein–Ligand Complex Prediction
Understanding protein–ligand interactions is fundamental to molecular biology and biochemistry. These interactions are at the heart of many cellular processes, from enzyme catalysis to signal transduction. This foundational knowledge of protein–ligand interactions has paved the way for structure-based drug design (SBDD), a pivotal area in pharmaceutical research.
In a study published on November 27 in Nature Methods, a research team led by Prof. ZHENG Mingyue from the Shanghai Institute of Materia Medica (SIMM) of the Chinese Academy of Sciences introduced a new deep-learning method, SurfDock. In various benchmarks, SurfDock has outperformed existing methods in docking success rates and adherence to physical constraints. SurfDock also exhibits remarkable generalizability to unseen proteins and predicted apo structures, achieving state-of-the-art performance in virtual screening tasks. In a real-world application, SurfDock identified seven novel hit molecules in a virtual screening project targeting aldehyde dehydrogenase 1B1, a key enzyme in cellular metabolism. This showcaseed SurfDock’s ability to elucidate molecular mechanisms underlying cellular processes.
SurfDock is a geometric diffusion network designed to generate reliable and accurate binding ligand poses. The diffusion process is conditioned on protein pocket and a random starting ligand conformation. Additionally, SurfDock incorporates an internal scoring module, SurfScore, which was trained on crystal protein–ligand complexes to estimate pose confidence. SurfDock integrates multimodal protein information, such as surface features, residue structure features and pre-trained sequence level features, into a surface node level representation.
As a result, SurfDock achieved top performance in docking success rates across several benchmarks, significantly outperforming previous deep learning (DL) methods in terms of the plausibility of generated poses. SurfDock also incorporated an optional force field-based relaxation step for protein-fixed ligand optimization, further improving its accuracy and validity. Moreover, SurfDock effectively generalized to new proteins, pockets and apo structures, while also being robust against varying ligand flexibility. In virtual screening (VS) scenarios, SurfDock not only matched but exceeded the performance of existing docking methods.
Finally, researchers showed the practical utility of SurfDock in a real-world small-molecule discovery project targeting aldehyde dehydrogenase 1B1 (ALDH1B1), where seven hit molecules with novel scaffolds were quickly identified. This performance, combined with its practicality and reliability, made SurfDock a valuable contribution to the SBDD community.
The ability to accurately predict protein–ligand complexes could significantly improve our understanding of protein biology and assist in designing new therapeutic agents. Researchers envision that SurfDock will become an essential tool in SBDD community with continual improvements, paving the way for chemical validation of novel targets important for fundamental biology and drug discovery.
DOI: 10.1038/s41592-024-02516-y
Link: https://www.nature.com/articles/s41592-024-02516-y
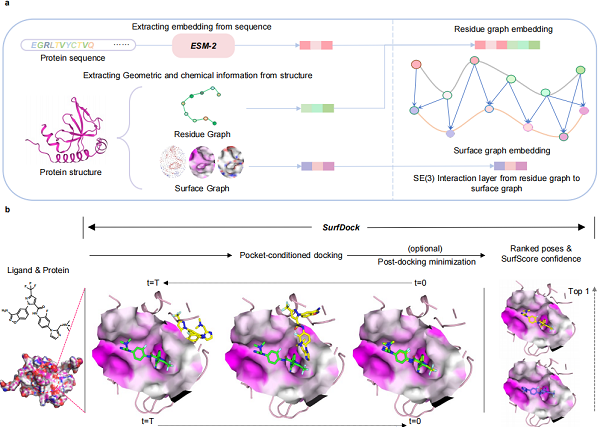
The overall architecture of SurfDock. (Image by ZHENG’s Laboratory)
Contact:
JIANG Qingling
Shanghai Institute of Materia Medica, Chinese Academy of Sciences
E-mail: qljiang@stimes.cn