


Study Decodes Cell Membrane Sphingolipid Remodeling: A Novel Therapeutic Pathway for advanced MASH
Metabolic dysfunction-associated steatohepatitis (MASH) is a chronic liver disease initiated by excessive lipid accumulation, triggering inflammation, hepatocyte ballooning, and fibrotic remodeling. While drugs targeting de novo lipogenesis can reduce hepatic lipid content, their efficacy in preventing MASH progression remains inconsistent. Emerging evidence suggests that specific toxic lipid species, rather than total lipid accumulation, drive MASH development. Sphingolipids, particularly ceramides, have been implicated in lipotoxicity, but the dynamic nature of sphingolipid metabolism and its potential as drug targets remain insufficiently explored.
In a study published in Cell Metabolism on February 26, a research team led by XIE Cen, LIU Hong, and LIU Yameng from the Shanghai Institute of Materia Medica (SIMM) of the Chinese Academy of Sciences (CAS), along with XIE Qing from Ruijin Hospital of Shanghai Jiaotong University School of Medicine, revealed disrupted sphingolipid metabolism in hepatocyte membranes as a key metabolic feature distinguishing hepatic steatosis from MASH, and identified sphingomyelin phosphodiesterase 3 (SMPD3) as the primary enzyme driving this process. SMPD3 activity, typically low in healthy livers, significantly increases during MASH progression in response to DNA damage signaling. Upon activation, SMPD3 translocates to membrane caveolae, disrupting membrane sphingomyelin-ceramide balance and promoting disease progression by enhancing caveolae-dependent lipid uptake and extracellular vesicle (EV) secretion from steatotic hepatocytes to exacerbate inflammation and fibrosis. Thus, SMPD3 emerges as a central hub in pathogenesis of MASH.
The research team analyzed samples from MASLD patients and MASH mice using metabolomics and public sequencing datasets. They discovered that ceramides in MASL stage primarily originate from de novo synthesis, while in MASH stage, ceramide production is largely driven by SMPD3-mediated sphingomyelin hydrolysis. Notably, membrane ceramide levels significantly increase during MASH progression. Hepatocyte-specific Smpd3 disruption did not affect baseline metabolism but prevented steatosis-to-MASH progression, while SMPD3 re-expression reversed this protection.
To further understand the role of SMPD3 in MASH progression, the team performed transcriptomic and proteomic analyses on livers and hepatocytes from multiple MASH knockout mice models. They found that SMPD3 localizes to cell membrane caveolae, where it regulates communication between hepatocytes, macrophages, and hepatic stellate cells via EVs. Caveolae, cholesterol- and sphingolipid-rich invaginations of the plasma membrane, play a crucial role in lipid uptake and regulation of lipid networks. EVs are secreted after fusion of late endosomes with plasma membrane, a process in which SMPD3 is essential. The team demonstrated that SMPD3 regulates endosome formation by altering sphingomyelin-ceramide balance at caveolae. Consequently, SMPD3 enhances lipid uptake through caveolar endocytosis and promotes EV release from steatotic hepatocytes. These EVs then play a key role in M1 macrophage polarization and hepatic stellate cell activation, driving inflammation and fibrosis.
The expression of SMPD3, typically low in healthy livers, increased significantly during MASL-to-MASH transition in patients. This study identified a lipotoxicity-DNA damage-SIRT1-SMPD3 axis as a critical signaling cascade MASH progression, with SMPD3 functioning as a sensor for lipotoxic stress. Administration of DPTIP, a selective SMPD3 inhibitor, significantly alleviated MASH in mice, shifting their hepatic transcriptional profiles from advanced fibrotic stages (F2-F3) towards earlier, pre-fibrotic stages (F0-F1) or even MASL. Since SIRT1 was found as a critical upstream regulator of SMPD3 in MASH, the researchers hypothesized that combining SIRT1 activation with SMPD3 inhibition could have synergistic therapeutic effects. By applying a dual-target design strategy and activity screening, they identified DC17, a bifunctional molecule that both activates SIRT1 and inhibits SMPD3. DC17 demonstrated superior efficacy in alleviating MASH compared to single-target interventions, suggesting that dual targeting the SIRT1-SMPD3 axis could be a promising therapeutic strategy.
This work established membrane sphingomyelin metabolic reprogramming as primary source of ceramide accumulation in MASH. Mechanistically, SMPD3 links lipotoxicity to MASH pathogenesis through its coordinated regulation of lipid uptake, inflammation, and fibrosis. The study proposed that SMPD3 inhibitors, especially dual-target compounds like DC17, could serve as promising therapeutic options for advanced MASH. These findings positioned SMPD3 as a potential therapeutic target for MASH, offering new possibilities for clinical intervention. It should be noted that, although ceramide accumulation is observed in the liver during MASLD progression, systemic sphingolipid dysregulation in metabolic diseases such as obesity and diabetes likely involves complex interactions between multiple organs. Therefore, while liver-specific sphingomyelin hydrolysis is critical in MASH, contributions from other organs must also be considered in broader metabolic context.
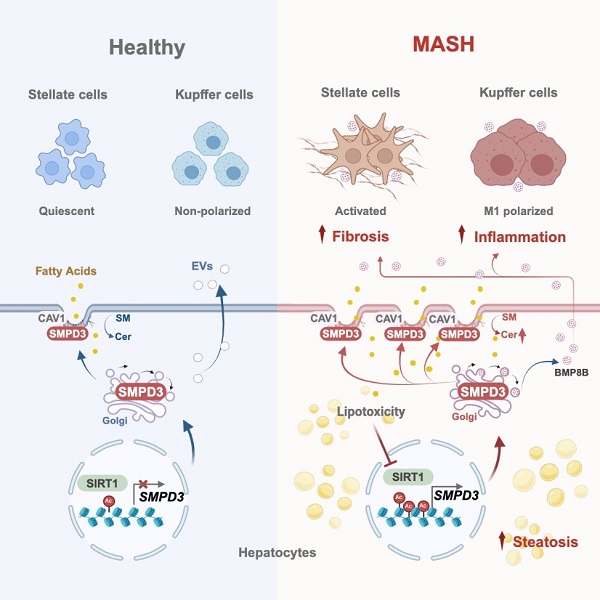
Illustration summary (Image by XIE’s Lab)
Contact:
JIANG Qingling
Shanghai Institute of Materia Medica, Chinese Academy of Sciences
E-mail: qljiang@stimes.cn