


PNAS|Cryo-EM Structures Reveal How GnRH Activates Its Receptor, Opening Pathways for New Therapies
In 1977, the Nobel Prize in Physiology or Medicine was awarded to Roger Guillemin and Andrew Schally for their discovery and synthesis of gonadotropin-releasing hormone (GnRH), a key regulator of reproductive function. Today, the GnRH receptor (GnRHR) remains at the forefront of biomedical research. GnRHR is a critical target for treating reproductive disorders such as infertility and prostate cancer. Leuprolide, a peptide analogue of GnRH targeting GnRHR, has dominated the market for therapies against prostate and breast cancers. However, the receptor’s low expression levels and instability have long hindered efforts to resolve its structure, leaving fundamental questions about how GnRH activates GnRHR unanswered.
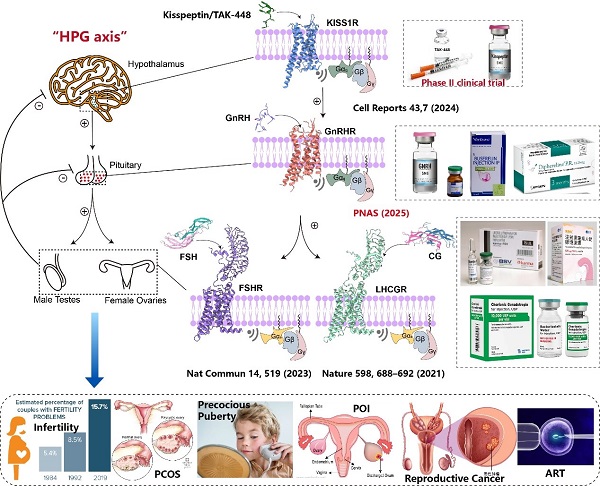
Figure 1. GPCR signaling pathways, protein structures, clinical drugs, and indications located in the hypothalamus-pituitary-gonadal (HPG) axis. (Image by Shen Shiyi)
In a study published in PNAS, a team led by DUAN Jia and XU Huaqiang (H. Eric XU) from the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences, reported high-resolution cryo-electron microscopy (cryo-EM) structures of GnRHR from two species: Xenopus laevis (African clawed frog) and Sus scrofa (pig, whose GnRHR closely resembles that of humans), each in complex with endogenous mammalian GnRH and Gq protein.
The team also analyzed nearly ten approved drugs targeting this receptor, shedding light on the conserved recognition of GnRH and the molecular basis of receptor activation. These results provide critical templates for designing next-generation therapies for reproductive diseases and cancers.
The researchers determined cryo-EM structures at 2.67 Å and 3.18 Å resolution for the frog and pig GnRHR-Gq complexes, respectively. GnRH was found to adopt a unique, conserved inverted “U-shaped” conformation, inserting both its N- and C-termini deep into the receptor’s orthosteric pocket. These termini interacted with key residues in transmembrane regions (TM3, TM5-TM7) and extracellular loops (ECL2 and ECL3). Comparative sequence analysis highlighted the high conservation of residues surrounding the binding pocket — including K3.32, Y6.51, and Y6.52 — that stabilize GnRH through hydrogen bonds and π-π interactions.
Functional experiments confirmed that mutations in these residues markedly impaired GnRH signaling. The study also showed that GnRH binding triggers conformational changes: the receptor’s N-terminus flips outward, and TM6 shifts to form the Gq binding interface.
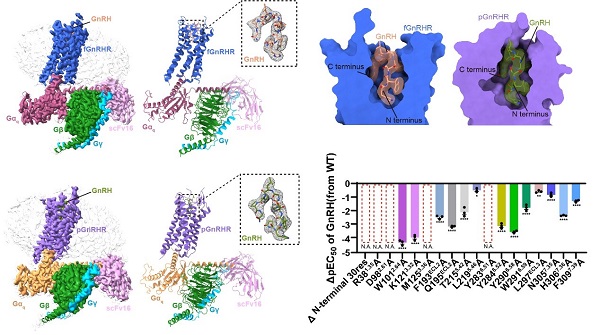
Figure 2. Cryo-EM structures of GnRH in complex with frog and pig GnRHR-Gq. (Image by Shen Shiyi)
By comparing active structures with inactive GnRHR bound to antagonists, the researchers identified key molecular switches driving receptor activation. Modeling and structural analyses of nine marketed GnRH analogues, such as triptorelin and nafarelin, revealed structure-activity relationships (SAR).
Importantly, they found that substitution of glycine at position 6 with D-amino acids (e.g., D-Trp) promoted a type II’ β-turn, pre-organizing the peptide for receptor binding and enhancing activity. This substitution also reduced the impact of mutations in pocket residues, offering clues for designing more robust peptide drugs.
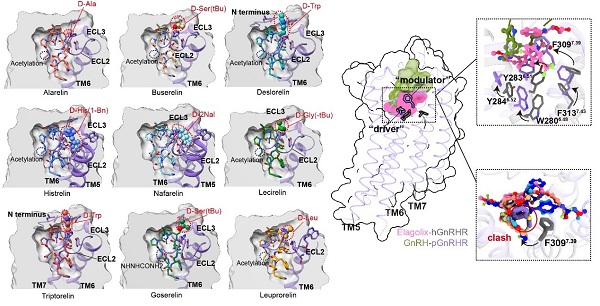
Figure 3. GnRHR drug modeling; GnRHR structure-activity relationship. (Image by Shen Shiyi)
The present study completes the structural map of key receptors along the hypothalamic-pituitary-gonadal axis, providing templates for rational drug design targeting reproductive disorders and related cancers.
DOI: 10.1073/pnas.2500112122
Link: https://www.pnas.org/doi/10.1073/pnas.2500112122
Contact:
DIAO Wentong
Shanghai Institute of Materia Medica, Chinese Academy of Sciences
E-mail: diaowentong@simm.ac.cn